Module 0
Setup
To follow this course, you will need to install a few things.
Shell
Windows
Enable Windows Subsystem for Linux (WSL)
- Winkey (⊞) + r to open Windows Run
- Type “control panel” and press Enter (↵)
- Click “Programs”
- Click “Turn Windows features on or off”
- Check boxes for “Virtual machine platform” and “Windows subsystem for Linux”
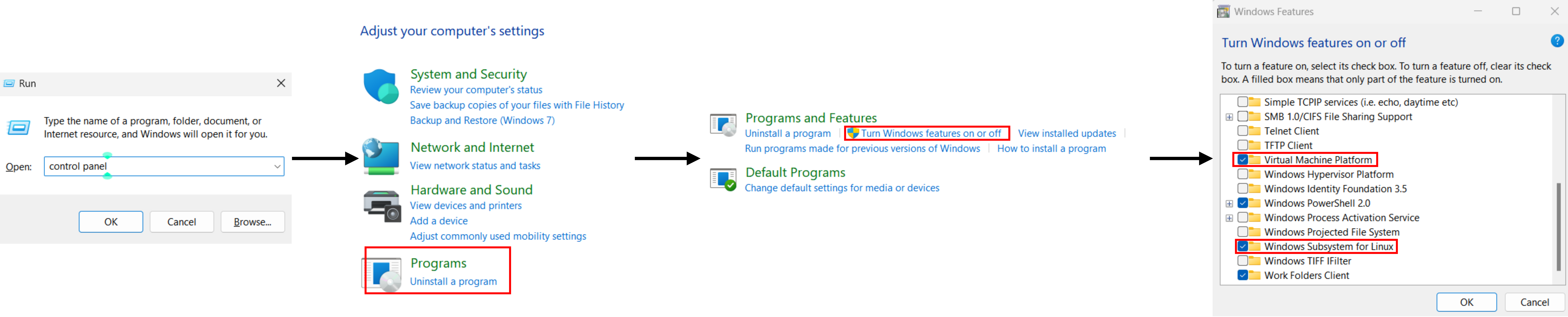
This might already be enabled depending on the exact version of windows. If that is your case you can move on to the next step
- Install a Linux distribution
- Winkey (⊞) + r to open Windows Run
- Type “powershell” and press Enter (↵)
- It should display a prompt ending in
>- For example:
PS C:\Users\username> >means the terminal is ready to receive a command
- For example:
- Type
wsl --updateto install the latest update if available - Then, once
>is visible again, typewsl --install -d Debian - This will install a distribution of Linux called ‘Debian’ (this will take up roughly 100 Mb of space on your computer)
- Once the installation completes, it will prompt for a username and password
- The password won’t be visible when you type. Don’t worry, that’s by design.
- Once entered, the Linux terminal will launch with a prompt ending in
$- Similar to
>in the Windows terminal,$means the Linux terminal is ready to receive a command
- Similar to
- Close the window
This is how it should look in your terminal:
PS C:\Users\username> wsl --update
Checking for updates.
The most recent version of Windows Subsystem for Linux is already installed.
PS C:\Users\username> wsl --install -d Debian
Installing: Debian GNU/Linux
Debian GNU/Linux has been installed.
Launching Debian GNU/Linux...
Installing, this may take a few minutes...
Please create a default UNIX user account. The username does not need to match your Windows username.
For more information visit: https://aka.ms/wslusers
Enter new UNIX username: your_username
New password:
Retype new password:
passwd: password updated successfully
Installation successful!
username@PCname:~$Here we installed Debian, but there are many other Linux distributions available. Ubuntu is the most popular, but we are using Debian because it is lightweight.
- Launch a dedicated Linux terminal
- Winkey (⊞) + r to open Windows Run
- Type “Debian” and press Enter (↵)
- A new Terminal window will open running Debian
- This will be the primary working environment
Linux
- Open the built-in terminal
- Press control + alt + T
MacOS
- Open the Mac terminal
- Press command + space to open the search bar
- Type “terminal” and press return
Installing Conda
Conda is a package management software that provides a convenient way to install other packages and their required dependencies, as well as manage virtual environments. For this workshop, we will install Miniconda, which comes with conda and a set of other essential packages.
The official Miniconda installation instructions can be found here - https://www.anaconda.com/docs/getting-started/miniconda/install
Feel free to navigate the above website and find the instructions appropriate for your system if you’re comfortable. If not, follow the instructions below.
- Open the terminal and create a new folder called “miniconda” in your home folder
mkdir ~/minicondamkdir: Command to make a new “directory”. “Directory” and “Folder” are functionally the same and can be used interchangeably.
~/: is the shortcut for your default home directory.- This can be replaced with the path to any other location on your computer if you want to create a new folder somewhere else instead.
- You can type
echo $HOMEand hit Enter to see the true path to your default home directory.
miniconda: name of the new directory we want to create.mkdir ~/miniconda: “Make a new directory called miniconda in the location ~/”
- Navigate to the newly created miniconda directory
cd ~/minicondacd: Command to “Change directory”, i.e navigate from the current working directory to a different directory.- You can type
pwdand hit Enter to see the path to the current working directory. - You can type
lsand hit Enter to list the contents of the current directory.
- You can type
~/miniconda: Path to the directory you want to navigate tocd ~/miniconda: “Change the current working directory to the location ~/miniconda`
- Select the installer:
Windows and Linux: Navigate to https://repo.anaconda.com/miniconda/, right-click Miniconda3-latest-Linux-x86_64.sh from the list and copy the link.
Windows users will still be using the Linux version as we will be working on WSL (see section Windows) instead of the Windows terminal.
MacOS: Navigate tohttps://repo.anaconda.com/miniconda/, right-click Miniconda3-latest-MacOSX-x86_64.sh if you have an INTEL Mac, or right-click Miniconda3-latest-MacOSX-arm64.sh if you have a M Series Mac, and copy the link.
Type uname -m in your terminal. If it returns x86_64, you have Intel. Otherwise, you have M series
- Download the miniconda installer; type
curl, and then paste the link you copied. For example, if you copied the “Miniconda3 Linux 64-bit” link, then the command will be
curl -O https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.shcurl: Command for retrieving data from URLs-O: Activates theOflag, which means the contents of the file in the specified URL will be saved with the same name- To see the list of all possible options and their explanations for curl, type
man curland hit enter. This will open the “Manual” for thecurlcommand.
- To see the list of all possible options and their explanations for curl, type
https://....: Link to the file to downloadcurl -O https://...- “Download the corresponding file from this URL”
This will download a file with the extension .sh, which signifies that it is a bash script.
- Execute the downloaded bash script. For example, if you downloaded the
Miniconda3-latest-Linux-x86_64.shfile, the commands will be:
bash Miniconda3-latest-Linux-x86_64.sh -u -b -p ~/miniconda/bash: Command to execute the bash scriptscript_name.sh: Name of the script to execute; in our case, the name of the script isMiniconda3-latest-Linux-x86_64.sh. You can then optionally specify flags unique to the script being executed. In the case of our script,-u: Activates the “Update” flag; updates an existing directory with new contents-b: Activates the “Batch” flag, which will execute the script without requiring any prompts from the user. You can run the above command without-bto see what that looks like.-p: Activates the “Prefix” flag; unlike the previous flags this one requires an input. Here, it is the prefix to be added to the new content that will be created. We specified~/miniconda/as the prefix, meaning all new files created by this script will be in the location~/miniconda
This would install conda and a number of other packages. Typing ls ~/miniconda/ will show you the new contents that have been created in the folder.
- Next, run the following to “initialise” conda:
~/miniconda/bin/conda initThis makes executing conda commands simpler and faster. Read more about initialization here:
Close and reopen the terminal.
Enter
conda -V; if the installation went correctly, you should see the conda version number. For example:
conda 25.7.0Setting up the conda environment
Next we will set up a conda virtual ‘environment’ - think of this as a box inside your computer where all the software you need for a particular project is contained. If you have multiple projects, you can set up multiple independent boxes that will not interfere with each other. This also allows you to simultaneously maintain different versions of the same software if you need to.
For the workshop, we have created a conda environment with many standard bioinformatics packages (available here). You can use the following command to load this environment onto your computer.
curl -O https://raw.githubusercontent.com/VishnuRaghuram94/NWUPathogenGenomicsWorkshop/refs/heads/main/NWU_2025_workshop.yml
conda config --set channel_priority flexible
conda env create -f NWU_2025_workshop.ymlThe environment can be activated with the following command:
conda activate NWU_2025_workshopOnce you have activated the environment, you will see the name of the environment in your bash prompt. It might look something like this
# Before activating
(base) username:~$
#After activating
(NWU_2025_workshop) username:~$Downloading example datasets
While the environment is active, download the setup script from the github page and run it. This will setup some folders and download some sample datasets which we will be using for the rest of this workshop. It will take some time, start the script and then feel free to grab a coffee! Once you see the prompt again, it means the script is done. The script will also output a “Setup successful” message.
This setup will take 4GB of space so make sure you have sufficient space available
curl -O https://raw.githubusercontent.com/VishnuRaghuram94/NWUPathogenGenomicsWorkshop/refs/heads/main/setup.sh
bash setup.shYou must activate an environment before you are able to use the tools within that environment. By default, you will be on the base environment. While it is fine to install packages while in the base environment, it can soon get crowded with multiple conflicting packages and lead to issues later down the line. Therefore it is good practice to keep the base environment as clean as possible while creating separate environments for different tools/projects. You can deactivate the current environment and go back to base using the command:
conda deactivate